MDR Medical Device Regulations – What Should the CUSTOMER Know?
Published 10.01.2025

The impact of the EU Medical Device Regulation (MDR 2017/745) on oral healthcare is significant. While there is extensive information available about patient and user safety and the manufacturer’s responsibility to develop patient-safe products, there is little information on how these regulations affect customers and decision-makers. Patient risks are reduced, but what about the customer’s risks?
Let’s provide some background. The Medical Device Regulation (MDR) is a mandatory EU legislation approved in April 2017. Manufacturers were given three years of transition time to prepare and comply with the requirements, with the deadline set for May 26, 2020. This timeline was tight for transitioning half a million different medical devices from the Medical Device Directive (MDD 93/42/EEC) to MDR. The COVID-19 pandemic, which began in 2020, led to an extension of the transition period to May 26, 2021. Until that date, new medical devices could be marketed under the old MDD framework. However, significant modifications to products were no longer possible unless the product was first approved under MDR, after which the changes could be notified and/or approved by a Notified Body.
This situation poses challenges, particularly for complex products like dental units, which require continuous development to meet market demands. Additionally, products under MDD could be sold until May 26, 2024, after which they must either be removed from the market or transitioned to MDR. Before this date, the EU Commission recognized that without an extension, there would be very few medical devices available on the market—a severe threat to patient safety, as medical devices must be replaced at the end of their lifecycle. The transition period for dental units (classified as risk class IIa) is extended until December 31, 2028.
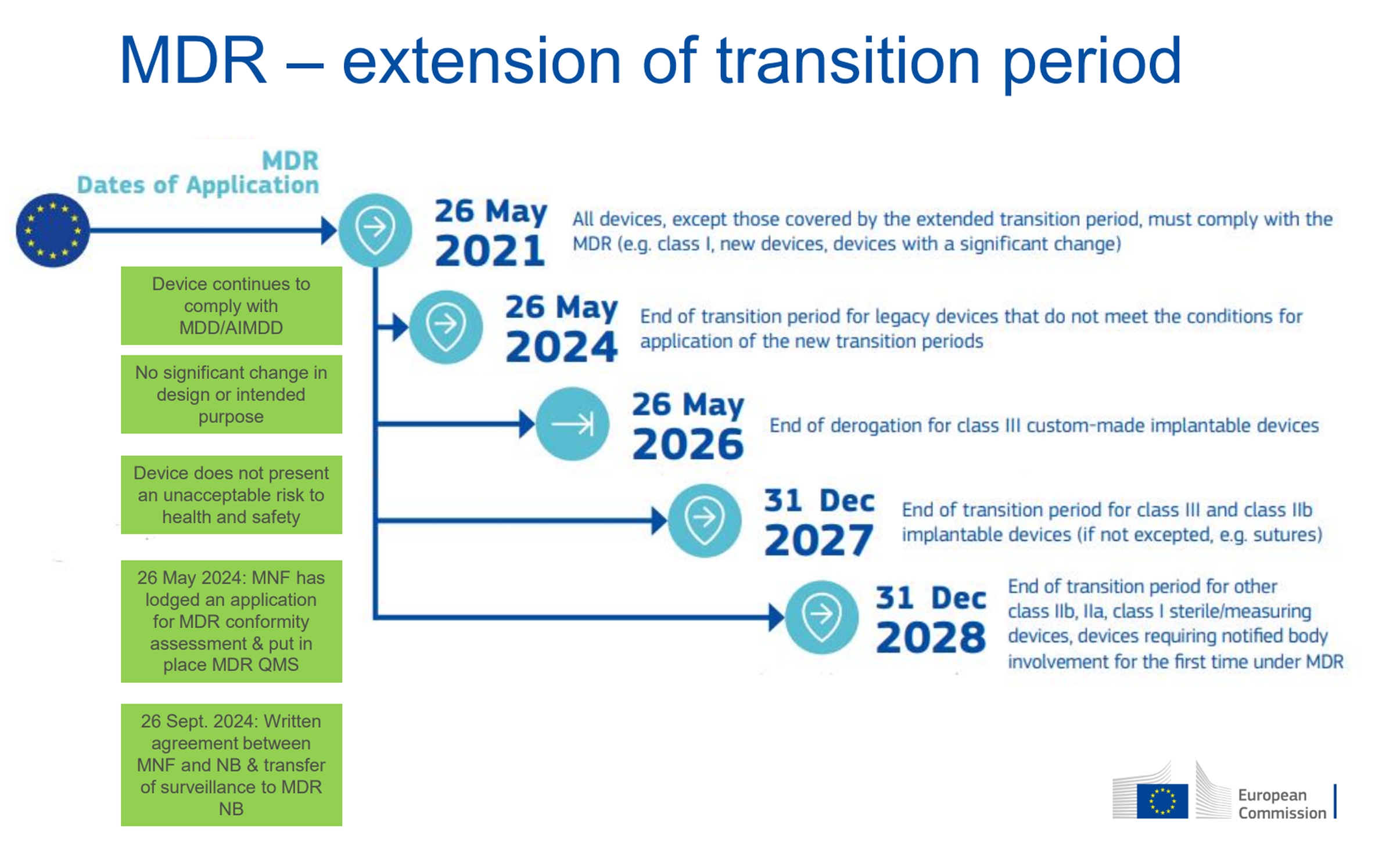
Why is the Transition So Lengthy for Medical Device Manufacturers?
The MDR demands significantly stricter risk analyses and extensive documentation compared to the outdated 1993 legislation. While large multinational companies are likely to manage the MDD-to-MDR transition, some products may be discontinued. For small and medium-sized enterprises, this transition is a matter of survival. Since May 26, 2021, these companies have had to freeze the development of their existing products under MDD. Transitioning to MDR requires substantial capital as development costs rise significantly.
What Should the Customer Understand?
A medical device, such as a dental unit, is purchased based on a justified need and typically has a lifecycle of 10–20 years, depending on the manufacturer. Purchasing a product under MDD means that it is already four years outdated. It will not receive significant updates or improvements through product development. This situation will not change until the product is transitioned to MDR, and the Notified Body has approved the EU certificate with its signature. At the same time, competitors’ products certified under MDR continue to develop annually to remain competitive.
If a medical device manufacturer fails to transition its products to MDR certification by December 31, 2028, the company will face the end of its business, as there will be no products to sell. The customer’s purchased device may still have years left in its lifecycle, but spare parts required for maintenance will no longer be available, and no one will answer calls for support when issues arise. In practice, the lifecycle of a dental unit would come to an end.
Risk and Compliance Challenges
It cannot be assumed that all companies always act strictly in accordance with laws, regulations, and good ethics. Sales must continue, and products must be delivered to customers. Product shortcomings and issues might be addressed to persuade customers to purchase, even though such actions may not be permissible under MDD certification.
A dental unit, classified under MDD as a class IIa device, requires conformity assessment and monitoring by a Notified Body. Notified Bodies conduct annual inspections to ensure compliance with regulations. These products are also subject to market surveillance. Customers may submit complaints, competitors monitor each other, and incidents posing risks to patients result in hazard notifications. Medical device manufacturers can be caught in these processes, and the Notified Body is responsible for ensuring product legality.
In cases of minor deficiencies affecting patient safety, the Notified Body may provide a timeframe, such as six months, to address the issue. However, significant factors affecting patient safety, such as electrical safety, could result in sales bans.
Complex Challenges with Changes
Challenges become significant when changes must be implemented under MDR certification that has not yet been obtained. For instance, can changes be reversed if key components of a device’s mainboard have been replaced or its functionality altered? A device’s display might need to be changed due to supply issues, and the new display might require additional EN60601 tests.
The worst-case scenario involves a Notified Body and/or authority ordering the withdrawal of products from the market. A medical device manufacturer may not financially survive a situation where products sold during the year must be recalled due to a fundamental product change that cannot be rectified. For the customer, this means paying for a product that cannot be used, with no response to complaints.
Learn more about MDR effects on business: Case of Dental Units.
MDR Effects on Business: Case of Dental Units – Fimet
Fimet Oy – A Reliable Finnish Medical Device Manufacturer
Fimet Oy is a Finnish medical device manufacturer established over 40 years ago. Fimet Oy holds the ISO13485 quality certification for medical device manufacturers.
Fimet’s products, Neo and F1 Arcus, are officially certified to meet the requirements of the MDR 2017/745 regulation.
This certification is evidenced in the EUDAMED database:
Devices/SPPs – EUDAMED
The certification process has been fully completed, as shown by the availability of Arcus & Neo product documentation in the official and public EUDAMED database:
Link to EUDAMED Database – Certificates
EUDAMED is the EU’s public database. Companies adhering to the law and prioritizing customer safety in patient safety are keen to report their information there. Fimet Oy’s submitted data is up-to-date and verified by the Notified Body (SGS Fimko). Customers can use this data as a benchmark to understand how a company’s information should appear when it complies precisely with the law to ensure safety for patients, users, customers, and the company itself.
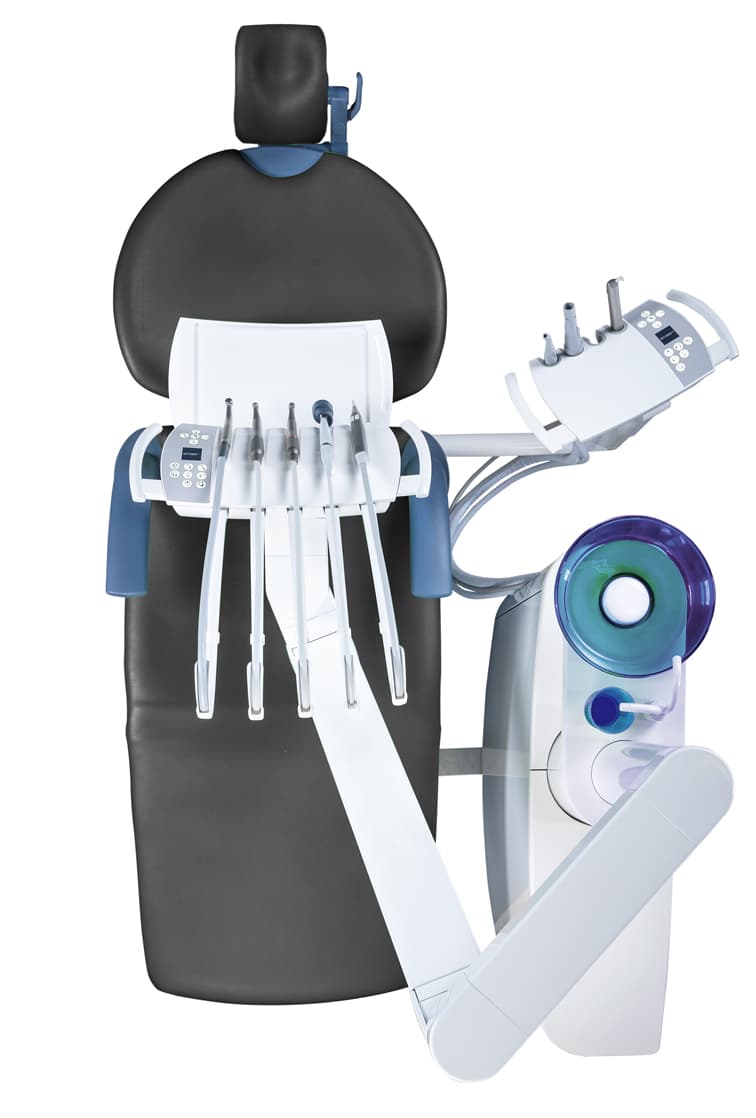
Arcus Dental Unit
With our newest development, F1 ARCUS, we are proud to present you with a high-performance unit that is ergonomic in its use and maintenance.
Learn More
Neo Dental Unit
The Fimet NEO dental treatment unit combines innovative solutions with elegant design. The treatment systems are ergonomic, high-quality units that can be customized for each user’s needs and preferences.
Learn More